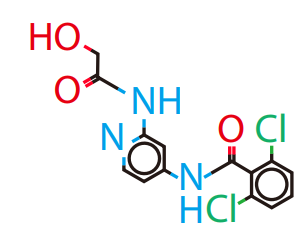
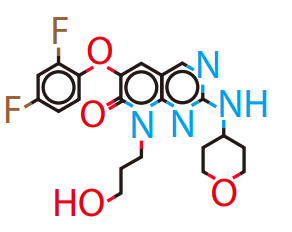
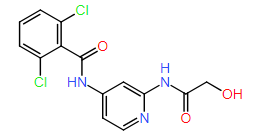
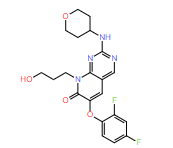
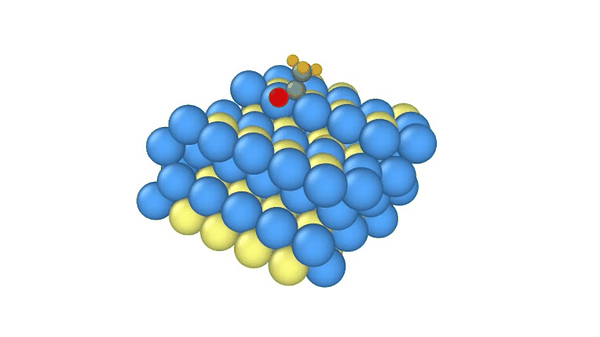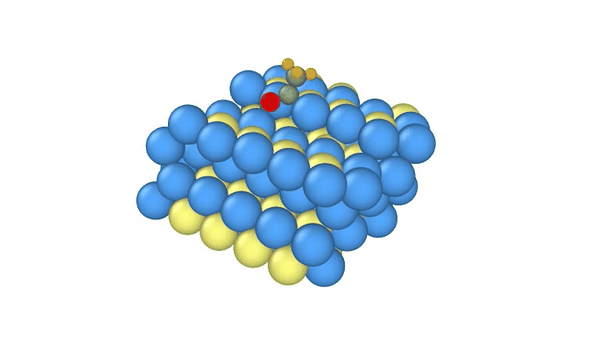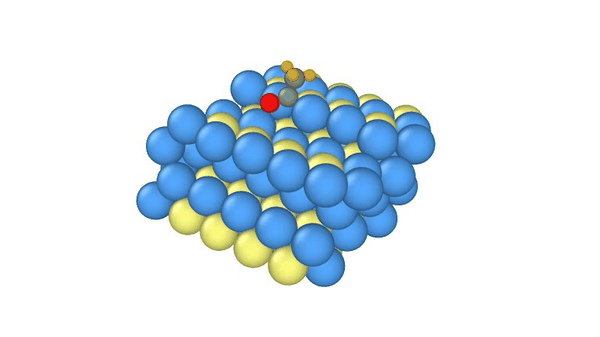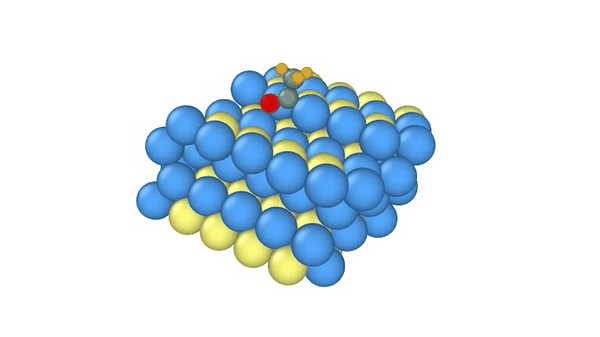
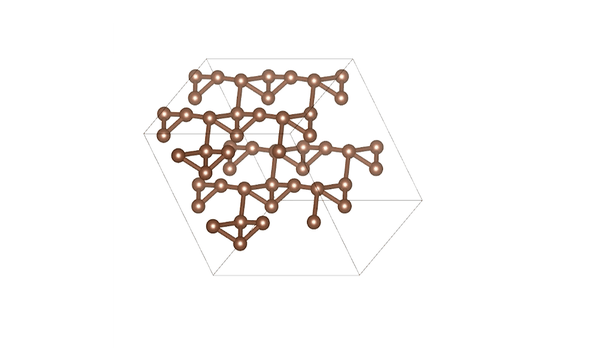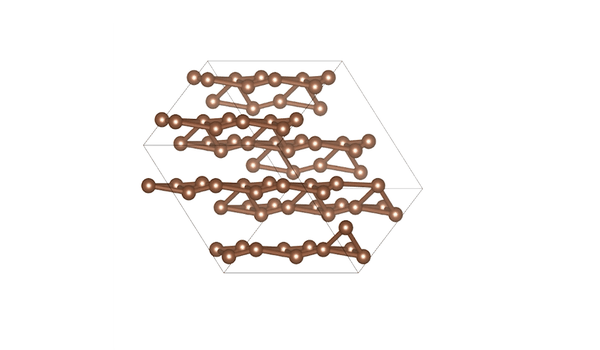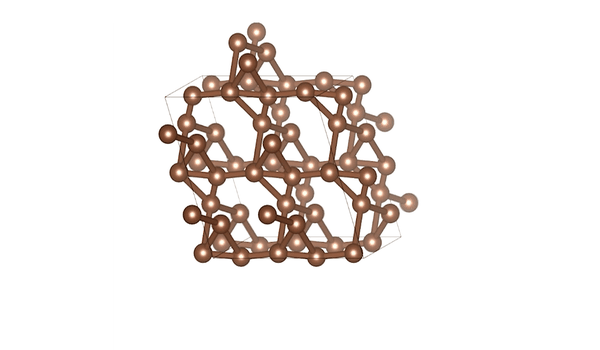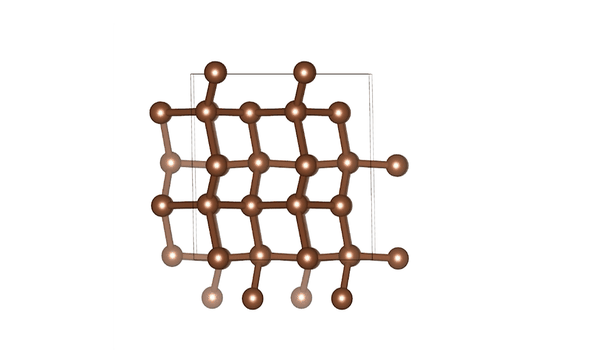
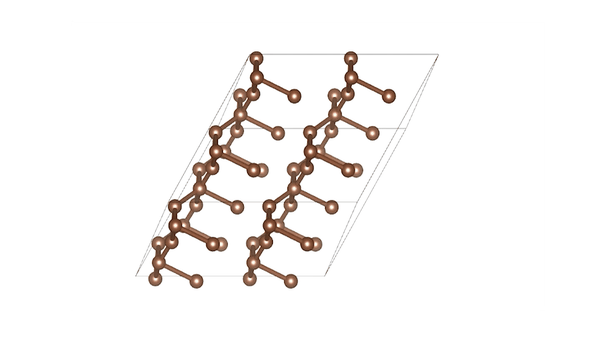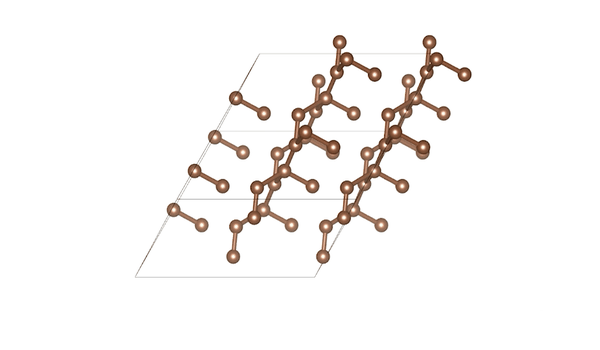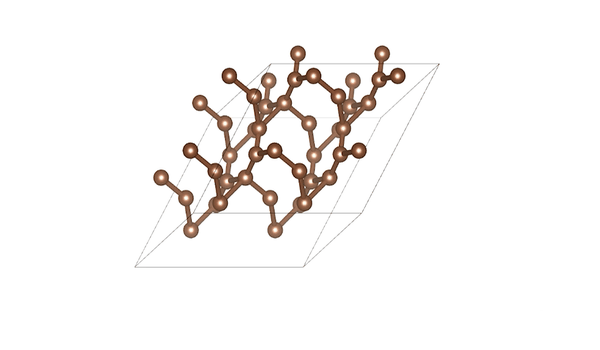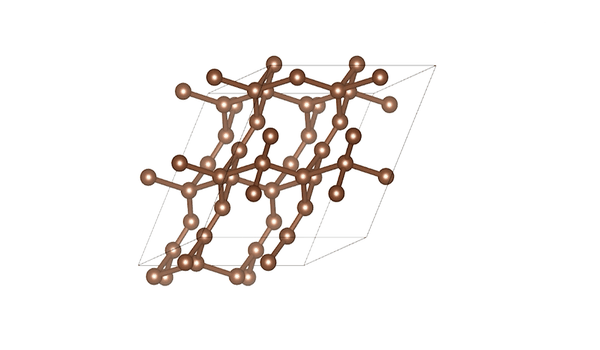
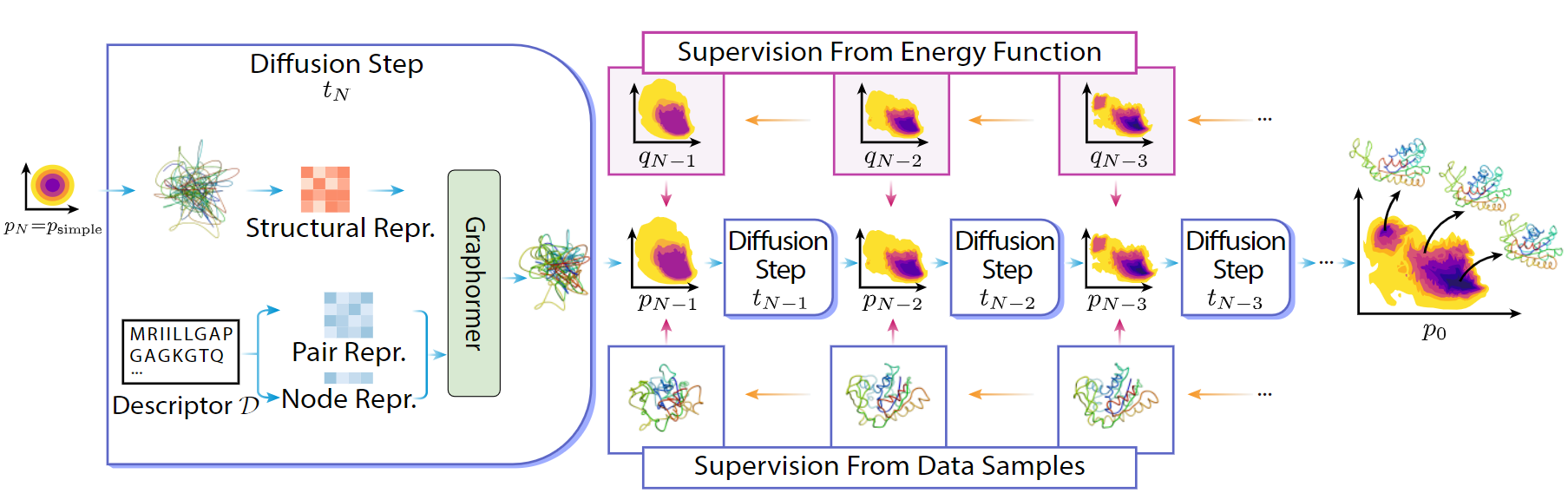
DiG generated structures for two proteins of SARS-CoV-2 viruses: the receptor binding domain (RBD) of the spike protein and the main protease (or 3CL protease). The contour lines show the distribution of millisecond molecular dynamics simulation results. Experimentally determined structures (from the protein databank) and the predicted structures by AlphaFold are shown in the 2D maps. DiG generated structures are mapped onto 2D maps as dots.
Slide the bar to check the conformational space coverage as more structures are being generated.